
【做计算 找华算】
超过1万个成功案例,全职海归技术团队、正版商业软件版权!
电化学二氧化碳还原(eCO
2
RR)能够将CO
2
直接转化为燃料和化学品,可以平衡大气碳平衡以及降低对化石燃料的依赖。CO是 eCO
2
RR 中最重要的 C1 砌块化合物之一。由于 对OCHO
*
中间体合适的吸附能,p 区金属,即 Bi、Sn、In 及其氧化物或络合物,有利于催化 CO
2
还原成甲酸盐。即使开发多种策略,例如合金化,掺杂等用于调制p区金属eCO
2
RR选择性,但是提高 p 区金属在 CO
2
-CO 转化的内在活性仍然存在挑战。由于独特的配位环境和电子结构,具有金属-氮共修饰碳(M-NC)结构的单原子催化剂(SACs)可用作在原子水平上理解构效关系的理想模型,并且定制催化的选择性。
因此,p区金属的原子化可以改良电子态,从而激活CO
2
-CO 的转化。铟(In)在eCO
2
RR领域收到广泛的关注。例如,无定形和高空位 InO
x
可以高选择性(高达 91.7%) 催化CO
2
RR 生产甲酸盐。另外,原子级分散的In 位点同样可以获得甲酸盐。例如,在改性碳基底(热解石墨、玻璃碳和硼掺杂金刚石)上固定的In卟啉 IX催化剂可以将 CO
2
还原为甲酸盐。
由于优先吸附 OCHO
*
中间体中的 O 原子,碳载体上卟啉类In-N
4
结构倾向于生成甲酸盐。然而,最新研究表明
In-N
4
位点催化剂可以在离子液体/MeCN 混合溶液中以 97% 的法拉第效率 (FE
CO
) 选择性催化 CO
2
-CO 的转化
。
在离子液体辅助下降低 CO
2
活化的反应势垒和 COOH
*
中间体的合适的吸附是CO高选择性的内因。相比,OCHO
*
中间体在 In-N
4
位点上的强吸附抑制甲酸盐的产生。不幸的是,在水溶液中 In-N
4
位点催化eCO
2
RR 的法拉第效率较低(FE
CO
= 53%)。

基于此,
中南大学刘敏、奥胡斯大学Kim Daasbjerg以及
山东大学胡新明等人
报道两种能够在水性介质中催化 CO
2
还原为 CO 的In SAC催化剂。光谱表征以及理论计算证实一种类型的 In SAC 具有空位、低配位的 In-N
3
-V 结构的特征,而另一种为典型的 In-N
4
配位结构催化剂。值得注意的是,
In-N
3
-V 位点表现出比 In-N
4
位点更高的内在 CO
2
-to-CO 活性
。
DFT 计算表明,In-N
3
-V 结构产生更高能量的 In电子态(主要是 s 和 p
z
轨道),有利于稳定 COOH* 中间体并促进 CO 的产生
。
与此同时,
In-N
3
-V以及In-N
4
位点催化CO
2
到甲酸的能垒更高,从而抑制甲酸盐的产生。
性能测试表明In-N
3
-V 位点可以在水溶液中实现 CO FE 为95%,而 In-N
4
位点只能达到 80%。
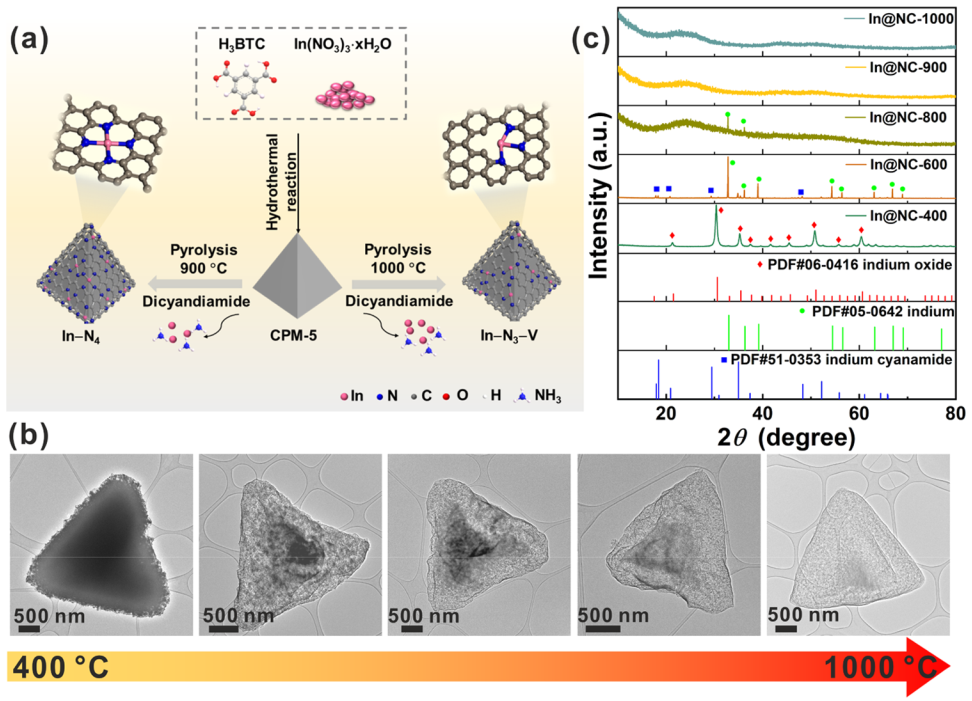
使用羧酸铟骨架结晶多孔材料(CPM)作为前驱体,在双氰胺存在下以及 Ar 气氛中高温碳化以合成In SAC催化剂,In@NC-
T
(T代表加热温度);XRD图显示当热解温度超过 900 °C,无法观察到与 In 有关的结晶峰,~26的峰来源于石墨碳,表明形成原子分散的In物种。
SEM图片显示In@NC-1000拥有更粗糙的表面;TEM和HRTEM观察到In@NC-1000的多孔结构,并且在整个碳结构中没有观察到纳米颗粒或者团簇的存在;球差矫正HAADF-STEM观察到孤立的亮点,证实In@NC-1000 中高度分散的 In 原子;ICP-OES确定In负载量为0.57 wt %;作为对比,In@NC-900的负载量为7 wt %。
图3. In@NC-900和In@NC-1000的光谱表征
1). In 3d XPS 谱,In@NC-900、In@NC-1000 和 In(III)四苯基氯化卟啉(InTPPCl)观察到相似的结合能,表明
In@NC-900 和 In@NC-1000 中 In 的平均价数为 +3
;在 In@NC-1000 中观察 In 3d 光谱向较低能量的偏移,表明 In 位点略高的电子密度;
2).
N 1s谱证实存在In−N(399.3 eV)结构
,以及吡啶 (398.0 eV)、吡咯 (400.7 eV)、石墨 (401.8 eV) 和氧化 (403.8 eV) N物种;
3). XANES谱显示In@NC-900 (27942.6 eV) 和 In@NC-1000 (27942.5 eV) 的吸收边位置接近In(III) InTPPCl (27942.6 eV) ,远离 In 箔 (27 940.0 eV),表明
两种催化剂的氧化态均为 +3
;
In@NC-1000 的吸收边能和白线强度略低于 In@NC-900,表明In@NC-1000 中的 In 电子密度相对较高
;
4). K边EXAFS谱,在 In@NC-900 和 In@NC-1000 中均未观察到 2.97 Å 处的 In-In 键,进一步证实
两种材料中单原子分散的In物种
;In@NC-900、In@NC-1000 和 InTPPCl 的
第一个壳层分别位于 1.63、1.68 和 1.64 Å的In-N 配位结构
;重要的是,In@NC-1000的In-N配位峰强度远低于In@NC-900和 InTPPCl,表明
I
n@NC-1000的低配位数。
图4. In@NC-900和In@NC-1000的配位结构表征
1). 基于拟合结果,
In@NC-900 的配位结构为In-N
4
,而 In@NC-1000中In与三个 N 原子和一个空位相邻,配位结构为In-N
3
-V
;
表明900°C的碳化温度导致四个 N 原子稳定的 In 原子,温度升高到 1000 °C,部分 In-N 键断裂,导致具有空位的不饱和 In-N 位点结构;
2). 计算的电荷密度空间分布表明In-N
4
中的 In 原子的缺电子特征,相反,In-N
3
-V中In原子保留更多电子,因此具备更丰富的电子环境。
图5. In@NC-900 和In@NC-1000的eCO
2
RR性能研究
1). 线性扫描伏安法(LSV)测试发现将 CO
2
吹扫到电解液中时,绝对阴极电流密度 |
j
| 会增加,并且In@NC-1000催化的电流密度增加更加显著;因此,
In-N
3
-V 位点比In-N
4
拥有更高的eCO
2
RR活性
;
在-0.87V vs RHE,In@NC-1000 催化的 CO部分电流密度 (|
j
co
|) 比In@NC-900更大,分别为 6.7 和3.1 mA cm
-2
;
2). 在电势范围在-1.07和-0.37 V之间,In@NC-1000 催化的CO
2
RR中 FE
CO
最大值为 95%,没有检测到甲酸盐;而In@NC-900催化的最大FE
CO
为80%,并且FE
formate
为~2%;
3). 进一步对比催化活性发现,在-0.87 V vs RHE电压下,In@NC-1000催化的eCO
2
RR的TOF = 5041 h
-1
,比 In@NC-900 对应的 TOF = 188 h
-1
高~27 倍;连续14h的CO
2
还原测试中,FE
CO
(≈95.1%) 和 |
j
| (≈2.3 mA cm
-2
) 在整个测试周期内保持稳定。
图6. DFT计算In SAC配位球对eCO
2
RR影响
1). DFT计算发现 In-N
3
-V 中In 轨道具备比In-N
4
更高的能量,因此更接近费米能级;并且在 In 原子的s 和 p
z
杂化轨道中更为明显;
2).CO
2
到 CO 的转化涉及两个电子和两个质子转移并形成 COOH
*
和 CO
*
中间体的过程;计算发现在In-N
3
-V 和 In-N
4
位点,第一质子耦合电子转移 (CO
2
*
+ e- + H+ → COOH
*
) 是速率决定步骤 (RDS)。
重要的是,在 In-N
3
-V 位点上 COOH
*
形成自由能为 0.64 eV,远低于 In-N
4
位点的1.48 eV
。
In-N
3
-V 通过降低吸附在 In 位点上的 COOH
*
的自由能促进 RDS,从而提高整体 eCO
2
RR;进一步,In-N
3
-V 中的 In s 和 p
z
轨道的上移有利于稳定 COOH
*
中间体,促进eCO
2
RR 形成 CO。
总之,使用含 In 金属有机框架辅助热解策略开发具有 In-N
4
和 In-N
3
-V 配位环境的单原子 In 催化剂;XANES以及XAFS证实当碳化温度为1000℃,饱和的In-N
4
结构会转化为不饱和的In-N
3
-V结构;相比于In-N
4
,In-N
3
-V催化的eCO
2
RR具备更高活性以及CO选择性;DFT 计算发现从 In-N
4
到 In-N
3
-V 的结构转变促使In s 和 p
z
轨道能量更接近费米能级,从而有利于稳定 COOH
*
中间体并促进CO的生成。
Simin Li, Xiuyuan Lu, Siqi Zhao,
et al
. p‑Block Indium Single-Atom Catalyst with Low-Coordinated In−N Motif for Enhanced Electrochemical CO2 Reduction. ACS Catal. 2022, 12, 7386−7395
https://pubs.acs.org/doi/10.1021/acscatal.2c01805?ref=pdf
